El síndrome de hipercortisolismo (código ICD 10) es un complejo de síntomas que se manifiestan bajo la influencia de una mayor síntesis de la hormona de la corteza suprarrenal.
La patología puede manifestarse a cualquier edad en cualquier sexo.
El síndrome se diferencia de la enfermedad en que en el segundo caso, el hipercortisolismo se produce de forma secundaria y la patología de la glándula pituitaria es primaria.
En medicina, existen tres tipos de hipercortisolismo, que se basan en la diferencia en las causas del inicio de la patología:
- exógeno
- endógeno;
- pseudo-síndrome.
En la práctica médica, también hay casos de síndrome de hipercortisolismo juvenil. El hipercortisolismo juvenil también se identifica como un tipo separado y se debe a cambios hormonales relacionados con la edad en el cuerpo del adolescente.
Exógeno
Bajo la influencia de causas externas, como el uso de fármacos que contienen glucocorticoides para el tratamiento, puede desarrollarse un hipercortisolismo iatrogénico o exógeno.
Básicamente, desaparece tras la abolición del medicamento que provoca la patología.
Endógeno
Los factores en el desarrollo del hipercortisolismo endógeno pueden ser las siguientes razones:
- (microadenoma hipofisario);
- tumores bronquiales;
- tumores testiculares;
- tumores de ovario;
- hinchazón o.
Un tumor provocador de los bronquios o las gónadas suele ser un corticotropinoma ectópico. Es ella quien provoca el aumento de la secreción de la hormona corticosteroide.

Síndrome de pseudo-Itsenko-Cushing
El hipercortisolismo falso se produce por las siguientes razones:
- alcoholismo;
- el embarazo;
- tomar anticonceptivos orales;
- obesidad;
- estrés o depresión prolongada.
La causa más común del pseudo-síndrome es una intoxicación grave por alcohol. En este caso, no hay tumores.
Factores de riesgo
De forma sintomática, el síndrome se manifiesta por los siguientes signos específicos:
- Obesidad, con un pronunciado depósito de grasa en la cara, cuello, abdomen. En este caso, las extremidades se vuelven más delgadas. El síndrome se caracteriza por una cara en forma de luna.
- Enrojecimiento malsano de las mejillas que no desaparece.
- Aparecen estrías cianóticas en el abdomen.
- Puede aparecer acné.
- Ocurre osteoporosis.
- Alteraciones en el trabajo del sistema cardiovascular, hipertensión.
Trastornos como la depresión o las migrañas prolongadas pueden ser tanto la causa del hipercortisolismo como sus síntomas. Además, el apetito por tal alteración del sistema endocrino a menudo se vuelve exorbitante.
Un paciente que padece el síndrome de Itsenko-Cushing se caracteriza por la presencia de pigmentación en lugares donde la piel a menudo se frota con ropa.

Juvenil
El hipercortisolismo en los niños ocurre debido a la hiperplasia de la corteza suprarrenal. Los síntomas de esta enfermedad pueden aparecer tan pronto como un año.
En presencia de síntomas característicos similares a los síntomas del síndrome en adultos, las siguientes manifestaciones ocurren en niños:
- susceptibilidad a enfermedades;
- escaso desarrollo de las habilidades mentales;
- desarrollo físico deficiente;
- cardiopatía.
Si la enfermedad se manifiesta antes de la adolescencia, puede comenzar la pubertad prematura. Si la enfermedad se manifiesta en la adolescencia, habrá un retraso en el desarrollo sexual.
Si un niño recién nacido muestra todos los signos de patología, entonces es muy posible que los tenga. En más del 80% de las enfermedades con síndrome de Itsenko-Cushing menores de un año, la causa es un tumor benigno de la corteza suprarrenal.
Entre mujeres
Las mujeres tienen 10 veces más probabilidades de desarrollar hipercortisolismo que los hombres. La principal categoría de edad de los enfermos es la mediana edad.
En las mujeres, se manifiesta en los siguientes síntomas:
- Aumento del crecimiento del vello en labios, pecho, brazos y piernas.
- Hay amenorrea, anovulación.
- El hipercortisolismo en mujeres embarazadas provoca un aborto espontáneo o un defecto cardíaco en un niño.
Las mujeres tienen más probabilidades que los hombres de tener formas graves de osteoporosis. De hecho, esta manifestación de la enfermedad puede conducir a formas graves de discapacidad incluso antes del inicio de la menopausia.
El síndrome de hipercortisolismo conduce a una disminución de la libido tanto en mujeres como en hombres. En este último, también se manifiesta por impotencia.

Tipos de hipercortisolismo
En la tipología del síndrome de Itsenko-Cushing, se distinguen dos tipos de patología: primaria y secundaria.
El hipercortisolismo primario se detecta en violación de las propias glándulas suprarrenales, con la aparición de un tumor funcional de la corteza. Tales neoplasias pueden ocurrir en otros órganos, por ejemplo, las glándulas sexuales.
El hipercortisolismo secundario se asocia con cambios en la glándula pituitaria, cuando las neoplasias en el sistema hipotalámico-hipofisario provocan un aumento hormonal.
¿Cómo puede continuar el síndrome?
La patología puede ser latente, con un ligero aumento de la síntesis hormonal y pronunciada.
Los médicos distinguen tres formas de manifestación de la enfermedad:
- Hipercortisolismo subclínico, ocurre en una etapa temprana o con pequeñas formas de tumores, que se manifiestan por aumento de la presión arterial, disfunción de las gónadas.
- Iatrogénico surge de los efectos de un fármaco para el tratamiento de enfermedades reumáticas, la sangre. Con el trasplante de órganos, se detecta en el 75% de los casos.
- Funcional o endógeno El hipercortisolismo se detecta con patologías graves de la glándula pituitaria, con diabetes mellitus. Los pacientes con adolescencia requieren una observación especial.
Hasta el 65% de los casos se deben a hipercorticismo iatrogénico.
Grados
Según la gravedad del curso de la enfermedad, se distinguen tres grados:
- Ligero con ligera obesidad, el estado normal del sistema cardiovascular.
- Medio con el desarrollo de problemas con las glándulas endocrinas, aumento de peso en más del 20% de su propio peso corporal.
- Grave con el desarrollo de complicaciones graves y obesidad grave.
De acuerdo con la tasa de desarrollo de la enfermedad y sus complicaciones, es posible distinguir: una forma progresiva (el período para el desarrollo de la patología de seis meses a un año) y una forma gradual (de 1,5 años o más).

Diagnóstico
Los siguientes métodos se utilizan para diagnosticar esta enfermedad:
- un análisis de sangre y corticosteroides;
- pruebas hormonales de orina;
- radiografía de la cabeza, huesos del esqueleto;
- Resonancia magnética o tomografía computarizada del cerebro.
El diagnóstico se hace claramente si todos los estudios están disponibles. Debe diferenciarse de la diabetes y la obesidad.
Tratamiento
Con el hipercortisolismo de diferentes formas, se requiere una terapia diferente:
- El hipercortisolismo iatrogénico se trata con abstinencia hormonal.
- Si se produce hiperplasia suprarrenal, se utilizan medicamentos para suprimir los esteroides, como ketoconazol o mitotan.
- Cuando ocurre una neoplasia, se utilizan un método quirúrgico y quimioterapia. En medicina, la radioterapia se usa para tratar el cáncer glandular.
Aplicar adicionalmente:
- diuréticos;
- hipoglucemiante;
- inmunomoduladores;
- sedantes
- vitaminas, calcio.
Si al paciente se le extirpan las glándulas suprarrenales, tendrá que hacerlo por el resto de su vida.
El método moderno de laparoscopia se utiliza en casos de adrenalectomía. Es seguro para el paciente y tiene un período mínimo de rehabilitación.
Complicaciones
En ausencia de tratamiento o el curso rápido de la enfermedad, pueden ocurrir complicaciones que ponen en peligro la vida del paciente:
- alteraciones en el trabajo del corazón;
- hemorragia en el cerebro;
- envenenamiento de la sangre;
- formas graves de pielonefritis con necesidad de hemodiálisis;
- lesiones óseas, incluida una fractura de cadera o una fractura de columna.
Se considera una condición que requiere una respuesta rápida. Conduce a un daño severo a los sistemas del cuerpo, así como al coma. A su vez, la inconsciencia puede provocar la muerte.
Pronóstico del tratamiento
La supervivencia y la recuperación dependen de.
Predice con mayor frecuencia:
- El porcentaje de muertes representará hasta la mitad de todos los casos de hipercortisolismo endógeno diagnosticado pero no tratado.
- Cuando se diagnostica un tumor maligno, sobrevive hasta 1/4 de todos los pacientes que lo tratan. De lo contrario, la muerte ocurre dentro de un año.
- Con un tumor benigno, la posibilidad de recuperación alcanza hasta 3/4 de todos los pacientes.
Los pacientes con una dinámica positiva del curso de la enfermedad deben ser controlados por un especialista durante toda su vida. Con observación dinámica y tomando los medicamentos necesarios, estas personas llevan una vida normal sin perder su calidad.
El hipercortisolismo es una enfermedad que se desarrolla cuando el sistema hipotalámico-pituitario se altera y tiene un curso multisintomático.
Esta dolencia fue descubierta y estudiada por dos científicos: el neurocirujano Harvey Cushing en Estados Unidos y el neurólogo Nikolai Itsenko en Odessa. En su honor, el hipercortisolismo también se denomina enfermedad de Itsenko-Cushing.
Las hormonas glucocorticosteroides son necesarias para el metabolismo en el cuerpo, pero cuando aumentan en el cuerpo, pueden ocurrir varios cambios.
¿Cómo se manifiesta esta enfermedad?
Los principales síntomas se manifiestan con mayor frecuencia:
- Obesidad;
- Aumento de la presión arterial;
- Debilidad muscular;
- Disfunción sexual;
- Pueden aparecer manchas de la edad en la piel;
- A las mujeres les crece vello en el pecho y la cara.
La obesidad en tales pacientes tiene un aspecto peculiar, es decir, el tejido graso subcutáneo se acumula en mayor medida en la región supraclavicular, hombros y vértebras cervicales, y también aumenta el abdomen. Como resultado de la redistribución del tejido graso, los brazos y las piernas se adelgazan y los músculos se atrofian. El rostro adquiere una forma de "luna", la piel se vuelve seca y escamosa, aparecen llagas difíciles de tratar y las mejillas tienen un color rojo púrpura. Otros síntomas incluyen estrías en el pecho, caderas, abdomen, que son rojas o moradas.
La manifestación más peligrosa en el hipercortisolismo es una violación del trabajo del corazón y los vasos sanguíneos, debido a esto, se produce el síndrome de hipertensión arterial. Con un aumento de la presión, aparecen dolores de cabeza, parpadeando "moscas" ante los ojos. Debido a la insuficiencia metabólica, se puede desarrollar diabetes mellitus, osteoporosis, que es muy difícil.
El hipercortisolismo conduce a una disminución significativa de la inmunidad, que sirve como catalizador para la aparición de úlceras, abscesos, pielonefritis, infecciones fúngicas de las uñas y la piel. Además, los síntomas incluyen una violación del sistema nervioso, puede haber trastornos del sueño, mal humor, psicosis.
En las niñas, después del inicio del ciclo menstrual, puede ocurrir amenorrea (una condición en la que no hay menstruación). Hay un retraso en el crecimiento y el desarrollo sexual, la voz se vuelve áspera.
¿Como resultado de qué puede desarrollarse la enfermedad?
Las razones de esta dolencia aún no se han identificado completamente, pero se ha establecido que la enfermedad de Itsenko-Cushing ocurre en mujeres diez veces más a menudo que en hombres.
Dicha enfermedad puede desarrollarse en cualquier rango de edad, pero con mayor frecuencia se enferman entre los 20 y los 40 años.
Las razones pueden ser diversas lesiones en la cabeza y daño cerebral (educación, inflamación), embarazo, neuroinfección, tumor de las glándulas suprarrenales, páncreas, pulmones, bronquios. La causa principal se considera un adenoma de la glándula pituitaria anterior.
¿Cómo identificar el hipercortisolismo?
 El médico debe examinar al paciente, interrogar y luego se realizan los diagnósticos de laboratorio. Con su ayuda, se determina la secreción diaria de cortisol en el torrente sanguíneo y la cantidad de cortisol libre en la orina diaria.
El médico debe examinar al paciente, interrogar y luego se realizan los diagnósticos de laboratorio. Con su ayuda, se determina la secreción diaria de cortisol en el torrente sanguíneo y la cantidad de cortisol libre en la orina diaria.
Para identificar la enfermedad del hipercortisolismo, debe realizar una pequeña prueba con dexametasona. Gracias a ella, puedes identificar un tumor pituitario.
Otro adenoma pituitario se determina mediante un examen de rayos X de los huesos del cráneo, una tomografía computarizada y una resonancia magnética del cerebro. Usando tales estudios de diagnóstico, es posible determinar la ubicación del tumor, su tamaño, crecimiento y con qué tejidos entra en contacto, lo cual es necesario para el nombramiento de su tratamiento correcto.
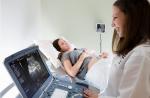
Además, es necesario realizar un estudio de las glándulas suprarrenales mediante ecografía, resonancia magnética y tomografía computarizada.
¿Qué determina la efectividad del tratamiento?
El hipercortisolismo puede desarrollarse rápidamente, es decir, todos los síntomas aparecen dentro de los 6-12 meses y puede haber un desarrollo gradual del cuadro clínico dentro de los 3-10 años. El tratamiento dependerá del diagnóstico correcto, la gravedad de la enfermedad y la velocidad a la que se desarrollan los síntomas. El tratamiento debe tener como objetivo eliminar las manifestaciones clínicas y normalizar los niveles de cortisol.
Con una gravedad moderada a leve, se usan medicamentos que evitarán que el cuerpo produzca un exceso de hormonas suprarrenales, o se prescribe radioterapia, que reduce la actividad de la glándula pituitaria. Si todo esto no da el efecto deseado, se utiliza un tratamiento quirúrgico. En el proceso de esta intervención, se extrae el tumor pituitario. O se realiza una adrenalectomía, es decir, la eliminación de una de las glándulas suprarrenales, pero después de dicha operación, se requiere una terapia de reemplazo constante.
Variedades de hipercortisolismo.
Funcional
El hipercortisolismo funcional se produce como resultado de diversas enfermedades que aumentan indirectamente el contenido de cortisol en el cuerpo. Tales dolencias incluyen: síndrome de ovario poliquístico, obesidad, cirrosis del hígado, hepatitis crónica, anorexia, trastornos del sistema nervioso, depresión, alcoholismo y otras causas de hipercortisolismo funcional son el embarazo y la pubertad.
Secundario
El hipercortisolismo secundario en mujeres embarazadas se desarrolla con un aumento de la hormona adrenocorticotrópica, que es responsable de las glándulas suprarrenales. Primero, se daña el hipotálamo y luego se afecta la glándula pituitaria y se desarrolla su tumor, y aparece un adenoma suprarrenal.
La sintomatología es muy similar a la del hipercortisolismo ordinario, el metabolismo está alterado, lo que puede causar un desarrollo y crecimiento deficiente del feto e incluso provocar su muerte. El diagnóstico se realiza sobre la base de anamnesis, examen, examen craneográfico de rayos X (la silla turca no aumenta mucho, ya que los tumores corticotrópicos no son grandes), resonancia magnética, se realizan exámenes de laboratorio de hormonas, se utilizan pruebas hormonales con dexametasona o metapirona.
Si una mujer quedó embarazada durante el período de tiempo en que tuvo una etapa activa de hipercortisolismo secundario, entonces se debe realizar un aborto. Para tener y dar a luz a un niño, la enfermedad debe estar en remisión, cuando la presión arterial es normal, el metabolismo no se altera y se realiza una terapia de sustitución. Solo el 30% de las mujeres con esta afección pueden tener y dar a luz a un hijo.
 Durante el embarazo, debe controlar constantemente la presión arterial, el peso corporal, el edema, los niveles hormonales, la micción y determinar la cantidad de azúcar en la sangre. Es necesario visitar a un endocrinólogo cada tres meses, seguir una dieta baja en sal y carbohidratos, consumir más frutas y vitaminas.
Durante el embarazo, debe controlar constantemente la presión arterial, el peso corporal, el edema, los niveles hormonales, la micción y determinar la cantidad de azúcar en la sangre. Es necesario visitar a un endocrinólogo cada tres meses, seguir una dieta baja en sal y carbohidratos, consumir más frutas y vitaminas.
Cuando un niño nace con bajo peso, con baja presión y con un aumento en la orina de las hormonas corticosteroides, el médico prescribe medicamentos glucocorticosteroides, el niño debe estar registrado con un endocrinólogo y un neurólogo.
Endógeno
El hipercortisolismo endógeno en el 80-85% de los casos se desarrolla con un tumor o hiperplasia hipofisaria. Sucede:
- Hipercortisolismo dependiente de ACTH;
- Síndrome ectópico de ACTH;
- Hipercortisolismo independiente de ACTH;
- Corticosteroma;
- Hiperplasia macronodular de la corteza suprarrenal;
- Hiperplasia micronodular de la corteza suprarrenal.
Las manifestaciones clínicas incluyen obesidad a una edad temprana, aumento de la presión arterial, osteoporosis, atrofia y debilidad muscular, cambios tróficos en la piel, amenorrea, crecimiento deficiente en los niños y aparición de tumores suprarrenales.
Subclínico
El hipercortisolismo subclínico se desarrolla en los tumores suprarrenales y ocurre en el 5-20% de las personas. Puede determinarse mediante un examen de hardware (ultrasonido, resonancia magnética, tomografía computarizada). Esta forma se detecta por casualidad, ya que no existen manifestaciones clínicas o son menos pronunciadas, el nivel de cortisol en la orina diaria se encuentra dentro de los límites normales. Pero si al paciente se le prescribe un tratamiento quirúrgico, se debe excluir el hipercortisolismo subclínico para que no surjan complicaciones postoperatorias.
Hipercortisolismo total.Etiología y patogenia del hipercortisolismo Dado que la corteza suprarrenal es un complejo formador de hormonas de mineralocorticoides, glucocorticoides y andrógenos, y dado que las hormonas esteroides se superponen parcialmente a los efectos biológicos entre sí, la patología del hipercortisolismo es muy mosaico. ACTH sirve como un regulador funcional para todas las zonas (para la zona del haz su papel es indiviso), y por lo tanto síndrome de hipercortisolismo total incluye hiperproducción incondicional de glucocorticoides, a menudo con síntomas más o menos pronunciados de hiperaldosteronismo e hiperandrogenismo.
Por etiología y patogenia del desarrollo. hipercortisolismo total existen las siguientes opciones:
YO.Suprarrenal primaria hipercortisolismocomo resultado de hiperplasia glandular primaria (independiente de ACTH) - síndrome de Itsenko-Cushing;
II.Secundario hipercortisolismo con estimulación excesiva de la glándula hipotalámica-pituitaria (dependiente de ACTH) - enfermedad de Itsenko-Cushing;
III.Secundario hipercortisolismo con producción ectópica excesiva de ACTH fuera de la región hipotalámica-pituitaria;
IV. Hipercortisolismo iatrogénicocon administración exógena de corticosteroides.
I. En una cuarta parte de los casos, el hipercortisolismo se asocia con una lesión tumoral primaria de la corteza. Esta patología se denomina independiente de ACTH. Síndrome de Itsenko-Cushing. Muy a menudo, este tumor crece a partir de las células de la zona del haz: glucosteroma (con exceso de glucocorticoides). Un tipo de glucosteroma es glucoandrosteroma con exceso de síntesis además de andrógenos. En este caso, la imagen del síndrome de Itsenko-Cushing se combina con hiperandrogenismo: en niños en forma de pubertad prematura, en mujeres, en virilismo.
Otra causa del síndrome de Itsenko-Cushing independiente de ACTH es hiperplasia no tumoral bilateral primaria de la corteza suprarrenal ... Ocurre en adolescentes y adultos jóvenes. El vínculo principal en la patogénesis se reconoce como un mecanismo estimulante autoinmune, similar a la enfermedad de Basedow. Se han obtenido experimentalmente inmunoglobulinas esteroidogénicas y mitosogénicas (crecimiento) para células de la corteza suprarrenal. En algunos casos, la hiperplasia no neoplásica bilateral primaria se considera una variante hereditaria autosómica dominante del síndrome: sinfotocomplejo de Carney. Una causa bastante rara de hipercortisolismo primario es hiperplasia bilateral de la corteza suprarrenal. Se cree que el mecanismo de este trastorno es la acción estimulante similar a la ACTH de un péptido gastrointestinal sintetizado por las glándulas gastrointestinales.
II. En la inmensa mayoría de los casos, la causa del hipercortisolismo es un tumor del lóbulo anterior de la glándula pituitaria (adenoma basófilo o tumores cromófobos que secretan ACTH en exceso). adrenocorticotropinomas ... Tal patología en Rusia se llama enfermedad de Itsenko-Cushing. Su patogenia se asocia a una mutación de la proteína G de las células pituitarias, que tiene afinidad por la corticoliberina, por lo que los adrenocorticotrofos adquieren una actividad excesiva frente a este factor de liberación hipotalámico.
Los métodos "antediluvianos" de tratamiento de la enfermedad de Itsenko-Cushing mediante el método de resección o extirpación de la glándula suprarrenal con adenomas pituitarios no reconocidos condujeron al rápido crecimiento de la misma. adrenocorticotropinomas debido a la estimulación de las células tumorales de la adenohipófisis con corticoliberina hipotalámica en el contexto de hipocorticismo, y la enfermedad de Itsenko-Cushing fue reemplazada por el síndrome de Nelson [crecimiento tumoral volumétrico en el cráneo sin signos de hipercortisolismo (si se resecaron las glándulas suprarrenales)].
III. Una causa relativamente rara de hipercortisolismo secundario son los tumores ectópicos de células del sistema endocrino difuso (apudomas), que secretan ACTH, con menos frecuencia corticoliberinas. Esta patología ocurre en cáncer de pulmón broncogénico, carcinomas del tracto digestivo, cáncer de tiroides medular, tumores de los islotes de Langerhans, timomas. Esta forma de hipercortisolismo a veces se combina con hipersecreción por células tumorales y otras sustancias biológicamente activas: vasopresina, oxitocina, gastrina, etc. De hecho, la patología descrita es el contenido del síndrome de crecimiento tumoral paraneoplásico. El nivel de ACTH en la secreción ectópica lo supera en la enfermedad de Itsenko-Cushing.
IV. Hipercortisolismo iatrogénico ocurre con el tratamiento a largo plazo con terapia a medio o corto plazo con dosis ultra altas de glucocorticoides.
Patogénesislas manifestaciones del hipercortisolismo total están determinadas por un exceso de hormonas de la corteza suprarrenal como resultado de la hiperplasia de los adrenocorticocitos.
Los glucocorticoides son hormonas del ciclo metabólico universal. Los estimuladores absolutos de su secreción son la ACTH, por lo tanto, el cuadro de hipercortisolismo está determinado por los efectos tanto de los corticosteroides como de la propia ACTH (por ejemplo, uno de los resultados de la acción de la ACTH puede ser la hiperpigmentación cutánea), así como la proopiomelanocortina y sus derivados. La combinación con las características del hiperaldosteronismo se explica tanto como consecuencia de la estimulación con ACTH como del efecto mineralocorticoide de grandes dosis de glucocorticoides. Recuerde que los mineralocorticoides son los reguladores más importantes del equilibrio de potasio-sodio y agua, y los andrógenos son reguladores de las funciones sexuales, el estrés y los procesos anabólicos.
Enfermedad de Itsenko-Cushing... Una disminución de la actividad de la dopamina y un aumento en el tono del sistema serotoninérgico del sistema nervioso central aumentan la producción de corticoliberina, ACTH y luego cortisol (cortisolismo secundario) debido a una violación de los mecanismos de retroalimentación. El hipercortisolismo no tiene ningún efecto inhibidor sobre las estructuras nerviosas centrales. La enfermedad se caracteriza no solo y no tanto por un aumento en la secreción de ACTH, sino por la estimulación de la producción de hormonas suprarrenales: cortisol, corticosterona, aldosterona, andrógenos.
Las violaciones de la relación hipotalámico-pituitaria se combinan con cambios en la secreción de otras hormonas tropicales de la glándula pituitaria: se inhibe la producción de hormona del crecimiento, disminuye el contenido de gonadotropinas y hormona tirotrópica, pero aumenta la secreción de prolactina.
La clínica de la enfermedad de Itsenko-Cushing está determinada por un trastorno de todo tipo de metabolismo, regulado por hormonas esteroides de las glándulas suprarrenales.
Violación metabolismo proteico En general, procede bajo el signo del catabolismo proteico principalmente en músculos y elementos mesenquimales (miocitos, células cutáneas, tejido conectivo, huesos, órganos linfoides), e incluso los procesos anabólicos predominan en el hígado y el sistema nervioso central. Por esta razón, se desarrolla miastenia gravis (debilidad muscular), atrofia muscular. La violación de la síntesis de proteínas se refleja en la composición de proteínas del tejido conectivo, glicosaminoglicanos, contenido de proteínas en el plasma sanguíneo (especialmente albúmina), inmunoglobulinas (anticuerpos). La desaminación mejorada de aminoácidos conduce a hiperazoturia. Se inhibe la colagenogénesis, lo que conduce al adelgazamiento y estiramiento de la piel en lugares de acumulación de grasa (síntoma del papel tisú), contribuyendo a la formación de estrías características (rayas de estiramiento) de color violeta-violeta por vasopatías, eritrocitosis e hipertensión. En pacientes jóvenes, se altera el crecimiento y el metabolismo de la vitamina D. Se inhibe la cicatrización de heridas.
Metabolismo de la grasa . Lo mas Xuna manifestación típica del hipercortisolismo es la obesidad de la localización central: en el contexto de la hipotrofia de las extremidades, la grasa se deposita en el abdomen, la cara, el cuello, en el espacio interescapular. Las causas más probables de obesidad son polifagia, hiperinsulinismo, distribución desigual de los receptores de insulina y glucocorticoides en varios lipocitos, estimulación de la producción de leptina por los adipocitos por los corticosteroides y efectos lipogenéticos directos de ACTH y glucocorticoides. Se observa un exceso de receptores de glucocorticoides en los lipocitos centrales, y el insulinismo potencia la lipogénesis en ellos, aumenta el suministro de glucosa y ácidos grasos.
Un exceso de glucocorticoides tiene un efecto lipolítico, provocando predominantemente hiperlipoproteinemia tipo II (por lipoproteínas de baja y muy baja densidad, colesterol, triglicéridos), que, por el mecanismo de desarrollo, puede atribuirse a las formas de producción y retención. El desarrollo de hiperlipoproteinemia se asocia con una mayor síntesis de triglicéridos en el hígado, lipólisis, bloqueo de los receptores apo-B de muchas células consumidoras.
Metabolismo de los carbohidratos ... Los glucocorticoides tienen un efecto contrainsular: inhiben el trabajo de los transportadores de glucosa (glucosa-4) en los tejidos dependientes de la insulina (lipocitos, miocitos, células del sistema inmunológico) a favor de los órganos no dependientes de la insulina: el sistema nervioso central, el corazón, el diafragma y otros. En el hígado se potencian la gluconeogénesis, la glucogénesis y la glucogénesis. En algunos pacientes con reservas insuficientes de células β del páncreas, se forma diabetes mellitus secundaria no insulinodependiente, complicada por cetoacidosis debido a la alta cetogenicidad de los glucocorticoides (que, por cierto, es típica de la diabetes mellitus insulinodependiente). En otros pacientes, en el caso de hiperfunción de las células β de los islotes de Langerhans, se desarrolla hiperinsulinismo, que estabiliza la situación, y no se produce una diabetes esteroidea manifiesta.
Metabolismo agua-sal y equilibrio ácido-base ... Se caracterizan por la retención de sodio y la pérdida de iones de hidrógeno y potasio, por lo que el contenido de K + en las células de los tejidos excitables (neuronas, cardiomiocitos, miocitos), así como en el plasma sanguíneo y los eritrocitos, se reduce significativamente. Se desarrolla alcalosis hipopotasémica. Los volúmenes de líquido extracelular y sangre (hipervolemia, pletora) aumentan. Se inhibe la absorción de calcio en el intestino y aumenta su excreción en los riñones. Se desarrollan nefrocalcinosis y nefrolitiasis, se une pielonefritis secundaria. El resultado puede ser insuficiencia renal. Una disminución del calcio en el cuerpo conduce al desarrollo de hiperparatiroidismo secundario. La hormona paratiroidea activa la transición de las células madre óseas a osteoclastos e inhibe la transformación de estas últimas en osteoblastos. El cortisol también inhibe la transición de osteoclastos a osteoblastos. Un aumento de los osteoclastos y un aumento de su actividad provocan la reabsorción ósea. Este último pierde la capacidad de fijar el calcio, como resultado de lo cual se forma la osteoporosis.
El sistema cardiovascular . El hipercortisolismo crónico causa hipertensión sintomática, cuyo desarrollo se asocia con los siguientes mecanismos:
1) un aumento del volumen sanguíneo (hipervolemia, pletora),
2) un aumento en la sensibilidad de los receptores adrenérgicos de los vasos resistivos a los factores presores debido a un aumento en el contenido de sodio y una disminución del potasio en los miocitos de los vasos resistivos (es decir, debido a un aumento en su tono vasomotor),
3) edema de músculos lisos de arteriolas y vénulas,
4) activación del sistema renina-angiotensina debido a la estimulación glucocorticoide de la síntesis hepática de α 2 -globulina (angiotensinógeno) y endotelina I,
5) el efecto inhibidor de los corticosteroides sobre la liberación de péptido natriurético auricular.
EN el sistema inmune Se forma inmunodeficiencia secundaria, insuficiencia fagocítica, que se manifiesta por una disminución de la resistencia a enfermedades infecciosas. Se desarrollan infecciones cutáneas bacterianas y fúngicas. Por este motivo, y por exceso de andrógenos, aparece el acné (acné vulgar) y la dermatitis perioral pustulosopapular.
Funciones sexuales. Una de las manifestaciones tempranas y permanentes de la enfermedad de Itsenko-Cushing es una violación de la función sexual, que es causada por una disminución de la función gonadotrópica de la glándula pituitaria y un aumento en la secreción de andrógenos por la corteza suprarrenal. En los hombres, se inhibe la producción de andrógenos por las glándulas sexuales (debido a la supresión de la secreción de gonadoliberina y hormona luteinizante por el mecanismo de control de retroalimentación), la libido disminuye y se desarrolla impotencia. Un exceso de andrógenos en el conjunto hormonal del hipercortisolismo en las mujeres forma hirsutismo (crecimiento excesivo del cabello), masculinización (adquisición de un tipo de cuerpo masculino), cambios en la conducta sexual, dismenorrea, amenorrea, abortos espontáneos, parto prematuro, infertilidad secundaria, virilización.
Sistema nervioso. El exceso agudo de glucocorticoides induce euforia, psicosis, alucinaciones y manía, mientras que el exceso crónico induce depresión.
Cambios en sangre . Los glucocorticoides estimulan la eritro y la leucopoyesis, desencadenan la apoptosis de linfocitos y eosinófilos, como resultado de lo cual se desarrollan eritrocitosis, neutrofilia, linfopenia, eosinopenia, cambian el estado de los sistemas sanguíneos coagulantes y anticoagulantes (desarrollo de trombohemorragia).
Hipercortisolismo parcial.Se debe a pronunciado el predominio de la secreción de un grupo de corticosteroides sobre los demás y está representado por los siguientes tipos:
1) hiperaldosteronismo (Primaria y secundaria);
2) síndrome adrenogenital (hiperandrogenismo).
Al mismo tiempo, prácticamente no existen formas parciales puras.
Hiperaldosteronismo primario (Síndrome de Connes).
I. La causa son tumores de la zona glomerular (aldosteroma) o con localización ectópica (ovario, intestinos, glándula tiroides). Un exceso de mineralocorticoides no inhibe la producción de ACTH, a diferencia del glucoster, por lo que no se produce atrofia de la parte sana de las glándulas suprarrenales.
II. Aldosteroma hereditario benigno con supresión de glucocorticoides.
III. Hiperplasia bilateral del área glomerular de la corteza suprarrenal de etiología desconocida. Como en el caso de la hiperplasia cortical micronodular, se discute el papel de los anticuerpos estimulantes en la etiología.
IV. Al comer raíz de regaliz (regaliz) y usar sus preparaciones, se altera la conversión de cortisol en cortisona (la presencia de ácido hiperricínico en materiales vegetales inhibe la enzima 11-β-hidroxilasa). En este caso, se reproduce el síndrome de pseudohiperaldosteronismo. Un defecto enzimático similar es la causa de la forma hipertensiva de hiperplasia suprarrenal hereditaria.
V. Síndrome de Lidl: pseudohiperaldosteronismo debido a hipersensibilidad del receptor primario a la aldosterona a niveles normales en sangre.
Vi. Administración iatrogénica de aldosterona.
En todas las formas de hiperaldosteronismo primario, la producción de renina, en contraste con las secundarias, es baja. La hipervolemia a través del mecanismo del receptor inhibe la síntesis de renina.
Hiperaldosteronismo secundario.Se desarrolla debido a la activación del sistema renina-angiotensina-aldosterona y avanza con un alto nivel de renina en el plasma sanguíneo. Las causas del exceso de secreción secundaria de aldosterona son:
1) Isquemia renal causada por daño a las arterias renales;
2) hipovolemia;
3) Hiponatremia y pérdida excesiva de sodio;
4) Hiperplasia primaria no tumoral de las células del aparato yuxtaglomerular del riñón ( síndrome de Bartter, un exceso de prostaglandinas E 2);
5) Reninomas (tumores de las células del aparato yuxtaglomerular del riñón);
6) Embarazo: los estrógenos estimulan la síntesis de renina y angiotensinógeno.
Patomorfología. Con el hiperaldosteronismo secundario, no hay tumor ni hiperplasia nodular, pero se observa hipersecreción e hipertrofia-hiperplasia difusa.
Las manifestaciones del hiperaldosteronismo consisten en síntomas típicos:
1) alteraciones de electrolito-agua - hipernatremia y retención de agua (hipervolemia), hipopotasemia y pérdida de iones hidrógeno.
2) hipertensión. Se acompaña de fluctuaciones ortostáticas (debido a la excreción de potasio, los barorreceptores pierden su sensibilidad a los cambios en la presión arterial sistólica y diastólica).
3) ausencia de edema - se compensa la producción de péptidos natriuréticos auriculares (atriopéptidos). Este mecanismo elimina parte del sodio y el agua e inhibe la formación de edema. La pérdida de potasio también se asocia con poliuria, principalmente por la noche.
4) hipopotasemia severa genera debilidad muscular, alteración del flujo de glucosa con flujo de potasio hacia la célula (efecto diabetogénico), "nefropatía hipopotasémica" con poliuria.
5) alcalosis - un desplazamiento del equilibrio ácido-base hacia el lado alcalino (en los túbulos contorneados distales, la reabsorción de Na + es a cambio de la liberación de K + y H +) se acompaña de hipocalcemia con posible tetania.
El eslabón principal en la patogenia.el hiperaldosteronismo secundario es una actividad muy alta del sistema renina-angiotensina-aldosterona, que avanza con hiperreninemia e hiperangiotensinemia severa, que están en relaciones antagónicas con péptidos natriuréticos. Por tanto, se forma una hipernatremia muy alta y un edema sistémico.
Síndrome adrenogenital.Se considera una secreción parcial excesiva de hormonas sexuales en las glándulas suprarrenales. (hiperandrogenismo ).
Las violaciones de la producción de hormonas sexuales de las glándulas suprarrenales son la causa de trastornos sexuales, conocidos colectivamente como: síndrome adrenogenital. Éstas incluyen:
1. Adquirido formas asociadas con varios tumores:
enfermedad y síndrome de Itsenko-Cushing , incluyendo glucoandrosteroma,
androsteromas ,
corticoestromas (se describen casos individuales en hombres).
2. Congénito formas. Forman parte de la estructura del síndrome adrenogenital llamado "Síndrome adrenogenital congénito" o (VDKN). La razón es la variedad de mutaciones genéticas que bloquean diferentes etapas de la esteroidogénesis determinada genéticamente.
Patogénesis. Síntomas típicos de la mujer hiperandrogenismo : hirsutismo, dismenorrea, virilismo y acné. En los niños, el tumor conduce a la pubertad temprana. El crecimiento de los niños se detiene. En las niñas, el síndrome congénito procede de acuerdo con el tipo heterosexual y forma pseudohermafroditismo, en los niños, de acuerdo con el tipo isosexual. En el 75% de los casos, el hipocorticismo se manifiesta y se acompaña de hiperpigmentación congénita de la piel, pérdida de sal en la orina (poliuria, hiponatremia, hipotensión muscular, hiperpotasemia, hipocloremia, acidosis, hipotensión), vómitos en fuente, antojos de alimentos salados. En el 25% de los casos, el hipocorticismo está latente.
En las mujeres se forma virilismo: hirsutismo, masculinización del físico, redistribución de la grasa según el tipo masculino, voz ronca, calvicie, atrofia de las glándulas mamarias, oligomenorrea y amenorrea, hipertrofia del clítoris, resistencia física, cambios en los estereotipos de conducta sexual. En los hombres, estos tumores no se reconocen. Tienen corticoesteromas bien reconocidos, formaciones malignas con producción de estrógeno mutante que causan feminización, ginecomastia, tipo de cuerpo y comportamiento femenino, hipotrofia testicular. Se requiere la mayor atención para las formas congénitas de síndrome adrenogenital con un bloqueo metabólico de la síntesis de cortisol en la dirección de los andrógenos. Hay muchas causas hereditarias de este tipo. Requieren distinciones de diagnóstico diferencial del hermafroditismo verdadero y falso de causas extraadrenales y no endocrinas y la determinación del sexo cromosómico. Las formas suprarrenales congénitas de hiperandrogenismo (síndrome adrenogenital) pueden ocurrir como parte del síndrome de hipocorticismo con síntomas de deficiencia de gluco y mineralocorticoides.
Se conocen las formas clásicas de hiperandrogenismo: virilizante más pérdida de sal pero sólo virilizante ... La forma no clásica se caracteriza por un inicio tardío de la enfermedad.
El eslabón principal en la patogénesis es el bloqueo enzimático de la conversión de 17-hidroxiprogesterona en 11-desoxicortisol, que conduce a la conversión excesiva de metabolitos en androstenediona. El hiperandrogenismo se desarrolla en el útero. Al mismo tiempo, se forma una deficiencia en la síntesis de mineralo y glucocarticoides. En este contexto, de acuerdo con el mecanismo de retroalimentación, aumenta la secreción de ACTH y se estimula el crecimiento de la corteza suprarrenal y la androsteroidogénesis. La corteza suprarrenal está agrandada debido a las zonas glomerular y reticular y se asemeja a la corteza cerebral. Clínicamente, el síndrome adrenogenital consta de dos síndromes – hiperandrogenismo e hipocorticismo , y principalmente en forma de hipoaldosteronismo.
Formas borrosas y claras [ "Hiperplasia (displasia) congénita de la corteza suprarrenal" ] ocurren hasta un 30%. Son la causa del hirsutismo y la adrenarquia. El hirsutismo es una razón de peso para buscar el síndrome del defecto de la 21-hidroxilasa. Un defecto en otras enzimas de la esteroidogénesis que crean un cuadro congénito del síndrome adrenogenital es extremadamente raro y se da en manuales especiales.
por Notas de la amante salvajeSíndrome de Cushing (hipercortisolismo) rara vez se diagnostica en hombres y niños. Es más probable que esta enfermedad afecte a mujeres en el grupo de edad de 25 a 40 años.
El desequilibrio hormonal causado por diversas razones conduce a cambios patológicos en el metabolismo, que se refleja en la apariencia.
La principal razón del desarrollo de la enfermedad de Cushing es exceso de producción de la hormona cortisol - un producto de la corteza suprarrenal. Y varios factores, que se enumeran a continuación, pueden contribuir a la interrupción del trabajo de este organismo.
Hipercortisolismo exógeno
Es causada por una sobredosis o el uso prolongado de medicamentos esteroides (recetados para el tratamiento del asma, la artritis reumatoide y en el período posoperatorio posterior al trasplante de órganos).
Hipercortisolismo endógeno

Es causada por un trastorno interno en el cuerpo. La disfunción hipofisaria (aumento de la producción de hormona adrenocorticotrópica) provoca la liberación de cortisol por la corteza suprarrenal. Las causas de la enfermedad pueden ser hiperplasia de la corteza suprarrenal y formaciones malignas, corticotropinomas. Lugares de posible localización: bronquios, ovarios, testículos.
Síndrome de pseudo Cushing
Los síntomas similares al hipercortisolismo pueden estar asociados con obesidad, intoxicación crónica por alcohol, embarazo, estrés, depresión y, en ocasiones, medicación anticonceptiva oral.
"Para reducir el riesgo de muerte y lograr resultados tempranos en el proceso de tratamiento, es recomendable buscar ayuda dentro de los primeros 5 años después de la aparición de la enfermedad".
Síntomas de la enfermedad de Cushing
1. Aumento de peso rápido y característico. Áreas de acumulación de grasa: cara (redonda y rubicunda), abdomen, región cervicotorácica. Al mismo tiempo, los brazos y las piernas se ven desproporcionadamente delgados.

2. Atrofia muscular de la cintura escapular y las piernas, acompañada de mayor debilidad y fatiga rápida.
3. Deterioro del estado de la piel: hiperhidrosis, aumento de la sequedad, tono marmóreo, adelgazamiento de la capa del epitelio superficial, pérdida de elasticidad (aparición de estrías) y funciones regenerativas (heridas que cicatrizan lentamente).
4. Disminución de la libido.
5. Crecimiento de cabello de tipo masculino en mujeres, falla y ausencia de menstruación.
6. Desarrollo de osteoporosis. En la etapa inicial, se distingue por dolor articular. En el futuro, puede manifestarse como fracturas espontáneas de las extremidades y las costillas.
7. Debido a los efectos hormonales negativos sobre el miocardio, surgen problemas con el funcionamiento del sistema cardiovascular. - cardiomiopatía, angina de pecho, hipertensión, insuficiencia cardíaca.
8. A menudo, el hipercortisolismo va de la mano de la diabetes mellitus esteroidea.
9. El sistema nervioso reacciona al desequilibrio hormonal con letargo, depresión, euforia, psicosis esteroidea.
Síndrome de Cushing: tratamiento

La enfermedad se diagnostica basándose en los resultados de análisis bioquímicos de sangre y orina. Además, el diagnóstico diferencial se lleva a cabo para identificar las causas: resonancia magnética de la glándula pituitaria, cavidad abdominal, radiografía capa por capa, estudio bioquímico de hormonas.
Al identificar las causas de la enfermedad de Cushing, se selecciona un método de tratamiento adecuado, destinado a eliminar la causa y restaurar el equilibrio hormonal.
Opción de medicación - el nombramiento de medicamentos que reducen la producción de cortisol.
Radioterapia - utilizado para influir en el adenoma hipofisario.
Intervención operatoria - se lleva a cabo con el objetivo de extirpar neoplasias de la glándula pituitaria y la corteza suprarrenal; en casos graves, se extirpan las glándulas suprarrenales y se prescribe terapia de reemplazo hormonal de por vida. Eficiencia: 70-80%, mejora rápida de la condición del paciente.
A menudo, en el tratamiento de esta enfermedad, se toman medidas complejas que combinan todos los métodos de tratamiento disponibles.